Mitochondrial Disease: Signs, Symptoms, Treatment, And Prognosis
Mitochondrial disease refers to a group of individual genetic disorders resulting from insufficient energy production in the body, reports Medscape. Mitochondrial disease develops when special compartments within our cells called mitochondria fail to produce enough energy. Any organ or system in the body can be affected since they all need energy to function. Muscles, kidneys, liver, ears, eyes, blood, and the hormonal system are most frequently affected. Close to 300 genetic mutations that cause mitochondrial disease have been identified, which can occur in either the DNA of the cell's nucleus or in the mitochondrial DNA.
Mitochondrial disease can lead to a wide variety of mild to severe symptoms, ranging from muscle weakness and learning disabilities to heart and neurological problems (via Cleveland Clinic). The diagnosis of mitochondrial disease is challenging, because there is no individual test that can determine whether or not a person has a mitochondrial disease. While mitochondrial disease is not curable, various treatments can minimize symptoms and help prevent worsening of the condition. These treatments include vitamins and supplements (e.g., coenzyme Q10, carnitine), as well as both aerobic and resistance exercise. While some people with mitochondrial disease lead fairly normal lives, others can experience severe adverse health effects in a short time.
Mitochondria can also malfunction as a secondary effect from another disease or condition. This secondary mitochondrial dysfunction (distinguished from primary genetic mitochondrial disease) can occur in people with Alzheimer's disease, diabetes, cancer, Lou Gehrig's disease, and muscular dystrophy.
What are mitochondria?
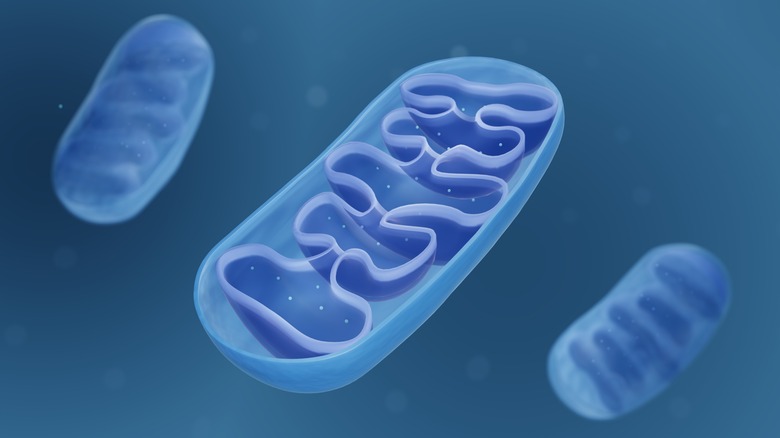
Mitochondria function as batteries within our cells to generate the vast majority of the energy we need (via Medical News Today). These tiny organelles (oval-shaped compartments) are enclosed by a double membrane consisting of an inner and outer membrane. ATP (adenosine triphosphate), the energy molecule of the cell, is produced in the inner mitochondrial membrane from the burning of food molecules together with oxygen.
The highest concentration of mitochondria (in the thousands) is in the cells of organs with high energy, like the heart and liver. Paradoxically, while mitochondria are necessary for a cell to live, they are also responsible for programmed cell death. This process, known as apoptosis, is used by the body to dispose of unwanted or abnormal cells (e.g., cancer cells). Mitochondria can also generate heat from non-shivering thermogenesis, and also play an important role in the cell's calcium balance by absorbing and storing calcium ions.
Mitochondria have their own genome, which is passed down from the mother only. These mitochondrial DNA are especially vulnerable to free radical damage because the mitochondria are a major site for the generation of highly reactive oxygen free radicals (ROS) as a spin-off of energy production. Moreover, unlike the DNA in the cell nucleus, mitochondrial DNA lack the ability to repair DNA damage. Mitochondrial DNA resemble bacterial DNA; this is related to the discovery that mitochondria evolved from primitive bacteria about two billion years ago (via Medscape).
Types of mitochondrial disease

According to the United Mitochondrial Disease Foundation (UMDF), hundreds of different types of mitochondrial diseases have been identified. Each condition is associated with a distinct array of symptoms, depending on which cells are affected.
For example, Leber hereditary optic neuropathy (LHON) is characterized by the sudden loss of central vision, beginning in one eye and affecting the other eye within a few weeks to months (via the UMDF). Vision loss results from impaired transmission of signals by the optic nerve to the brain. People with this condition are legally blind but never experience total blindness, since they continue to have some peripheral vision. LHON is caused by different variants of a mitochondrial mutation that leads to vision loss in some people, but not in others. Environmental factors, particularly smoking and heavy alcohol use, may increase the likelihood of vision impairment. LHON is more common in males, and occurs most often between the ages of 14 and 26.
Another mitochondrial disorder known as Leigh's disease typically begins within the first two years of life. Infants with this condition may have delayed development and other symptoms affecting the brain and nervous system such as ataxia (lack of muscle control) and seizures. While children with milder forms of the disease may live into their early 20s, others may succumb to respiratory failure in childhood. Two other mitochondrial disorders – Kearns-Sayre syndrome and chronic progressive external ophthalmoplegia (CPEO) – cause weakness of the eye muscles (via the Merck Manual). Other signs of these conditions include drooping eyelids, ataxia, deafness, and cardiomyopathy.
How mitochondrial disease affects the body

Mitochondrial disease can be likened to a power outage whereby the mitochondria in the body's cells cannot generate sufficient energy to fuel normal functioning of the cells (via a 2018 review in Peer J). As a consequence of this energy shortage, mild or severe disease and even death can occur.
The cells of tissues with the highest energy demands — muscle, heart, and brain — contain more mitochondria, and are thus more likely to be affected by defective (mutant) mitochondria. Diseases that affect these organs are termed mitochondrial encephalomyopathies. The development of disease is also determined by the proportion of defective mitochondria within the cell. Disease becomes apparent when, after a cell divides several times, the proportion of defective mitochondria in the cell surpasses 80-90%.
A deficit of cellular energy is not the only way that mitochondrial disease harms the body, notes the Muscular Dystrophy Association. Unused fuel molecules as well as oxygen can stockpile in cells packed with defective mitochondria. When producing energy (ATP), these dysfunctional mitochondria are unable to utilize oxygen efficiently to burn the unused fuel molecules. As a result, potentially toxic by-products such as lactic acid build up and cause lactic acidosis. This excess acid in the cell is linked to muscle fatigue and possibly impairment of muscle and nerve tissue. At the same time, unused oxygen can drive the excess production of toxic molecules known as reactive oxygen species (ROS), specific types of tissue-damaging free radicals.
Prevalence of mitochondrial diseases
In the United States, mitochondrial disease affects up to two million people, and occurs in 1,000 to 4,000 births each year (via a 2018 review of studies in Peer J). Some people are carriers of mitochondrial mutations, but never develop the illness. In Australia, for example, roughly one in 250 people carry a mitochondrial genetic defect whereas the risk of getting mitochondrial disease is around 1 in 5,000.
Determining the exact incidence of mitochondrial disease is challenging, because estimates may be disproportionately represented by cases of severe disease. People with mild or subtle symptoms of mitochondrial disease may be overlooked. In addition, according to the United Mitochondrial Disease Foundation, misdiagnosis of mitochondrial diseases occurs often. Though the disease occurs most often in childhood, adult onset is on the rise. Regardless of age, a person with mitochondrial disease is at special risk of severe complications during stressful periods, e.g., during and following an infection, starvation, dehydration, surgery, anesthesia, and intravenous antibiotics.
Causes of mitochondrial disease

Mitochondrial diseases are caused by mutations in the DNA found in the cell nucleus or in the much smaller amount of DNA located in the mitochondria (via the United Mitochondrial Disease Foundation). These gene mutations may either be inherited or occur randomly. Since genes provide the cell with instructions for making proteins, mutated genes produce proteins and RNA molecules (needed to assemble proteins) that function abnormally. In the mitochondria, the proteins and RNA involved in energy production in the form of ATP malfunction, resulting in an energy shortage. While a specific mitochondrial disease can be caused by different gene mutations, the reverse can also occur, whereby clinically different diseases can be triggered by the same mutation.
According to the Cleveland Clinic, three types of inherited gene mutations in mitochondrial disease have been identified. In autosomal recessive inheritance, the child inherits the disease when both parents pass a mutated gene onto their child. The probability of each child in the family developing the disease is 1 in 4. In contrast, an autosomal dominant pattern of inheritance requires that only one parent pass on a mutated copy of a gene to the child. The odds of each child in the family getting the disease increase to 1 in 2. A third way in which a mitochondrial disease is inherited involves mutations in the mitochondrial DNA. Uniquely, mitochondrial gene mutations pass only from mother to child. In mitochondrial inheritance, all the children in the family will inherit the disease.
Signs and symptoms of mitochondrial diseases

Sine nearly every cell in the body contains mitochondria, mitochondrial disease can affect many different organs and systems in the body and cause a wide assortment of symptoms.
The brain, heart, and muscles consume the most energy, and are thus impacted the most by mitochondrial disease. Disease that strikes the brain may cause developmental delay, autism, and learning disabilities in children. Other brain-related symptoms include migraines, dementia, seizure, and stroke. Nerve involvement can lead to fainting, pain, and intolerance to heat or cold.
Since hundreds of muscles are spread throughout the body, symptoms resulting from disease afflicting muscle tissue are very diverse, ranging from weakness and fatigue to constipation and diarrhea. Cardiomyopathy can occur when the heart is affected whereas diabetes can develop if the pancreas is involved. Mitochondrial disease of the liver or kidney may result in failure of these organs. Some types of mitochondrial disease can cause hearing loss, vision loss, or atrophy of the optic nerve. Systemic involvement is associated with short stature, weight gain difficulties, vomiting with no clear cause, and breathing difficulties.
Diagnosis
Diagnosing mitochondrial disease is tricky because it shares many symptoms with other illnesses (via the National Institute of Neurological Disorders and Stroke). A diagnosis of mitochondrial disease is more likely to be considered when more than three organ systems malfunction.
The diagnosis should begin with a medical and family history, along with physical and neurological exams that include assessments of strength and endurance as well as speech, vision, reflexes, and cognitive function. Various laboratory tests may be performed to detect diabetes and abnormalities related to liver and kidney function. The presence of lactic acidosis — common in mitochondrial disease — is indicated by high levels of lactic acid in the blood. Lactic acid can also be measured in the urine and cerebral spinal fluid.
Problems with the rhythm or pumping action of the heart can be spotted with an EKG, while brain health is evaluated with imaging scans such as CT and MRI. A biopsy of muscle tissue may be used to examine mitochondrial enzymes and mitochondrial proteins. The sample of muscle tissue may also show ragged red fibers, a sign of excessive defective mitochondria. Diagnosis of mitochondrial disease can be confirmed by a positive result from testing for genetic mutations in DNA samples from blood or muscle tissue. However, a negative test result does not rule out mitochondrial disease since some mutations cannot be identified.
Treatment
While mitochondrial disease has no cure, supportive treatments can help reduce symptoms and increase energy production in the mitochondria (via a review in Peer J). Some patients benefit from taking various nutritional supplements, alone or combined into a "mito cocktail." Creatine, a popular sports supplement,links with phosphorus in the body to form an energy molecule called phosphocreatine. This compound fuels intense but brief bursts of physical activity that cannot utilize the more slowly generated ATP (energy) from mitochondria, allowing ATP to be conserved in muscle cells.
Carnitine is another naturally occurring compound that plays a role in cellular energy production. It transports fatty acids into the mitochondria to be burned for fuel by all body tissues excluding the brain. Coenzyme Q10 (CoQ10) is a critical energy molecule that is deficient is some types of mitochondrial disease. CoQ10 helps convert food into energy as a part the mitochondrial electron transport chain that shuttles electrons to charge the "battery." CoQ10 absorption is maximized by using the ubiquinol form of the nutrient. Vitamin B1 and B2 (cofactors essential for the functioning of proteins within the electron transport chain) may also be helpful, notes Medscape. Since mitochondrial disease is associated with increased oxidative stress, antioxidants (vitamins C and E, lipoic acid) help neutralize destructive free radicals inside the cell.
If a person with mitochondrial disease can tolerate exercise, it can provide substantial benefits as well. Both aerobic and resistance exercise drive the creation of new, healthy mitochondria, while also decreasing the rate of mutation formation in the mitochondrial DNA.
Diet and mitochondrial disease

Nutritional support plays an essential role in the treatment of mitochondrial disease (via an article in the European Review for Medical and Pharmacological Sciences). In experiments in fruit flies, a diet with a low protein/carb ratio yielded less mitochondrial energy production than a diet with a higher protein/carb ratio.
Since the ketogenic/keto diet is very low in carbohydrates, its use as a nutritional intervention for people with mitochondrial disease has been proposed. The keto diet is neuroprotective and anti-inflammatory, and has been proven to reduce seizures in drug-resistant epilepsy. However, due to potential adverse effects, evidence suggests that the diet should be restricted only for children with types of mitochondrial disease that are manifested primarily by drug-resistant epilepsy.
Caloric needs for people with mitochondrial disease are based on guidelines established for other neurological disorders. Depending on physical activity, this amounts to around 30 calories per kilogram of body weight. Though not a part of the guidelines, a high protein intake of 1.0 – 1.5 grams per kilogram of ideal body weight is also suggested (excluding people with kidney failure). This amount of protein is higher than the RDA of 0.8 grams per kilogram of body weight and may help prevent loss of muscle mass. The texture of foods and beverages may need to be adjusted in individuals with swallowing difficulties. Tube feeding delivered directly to the gut may be warranted in cases of malnutrition or dehydration. Lastly, fasting should be avoided, as it may cause further fatigue and increase the risk of metabolic acidosis.
Prevention of mitochondrial disease
According to a 2020 review article in the journal Cell, a new technique has been developed to help prevent the transmission of genetic defects in families with mitochondrial disease. Known as "mitochondrial donation," the technique is designed to prevent the transmission of mitochondrial DNA from the mother to the child. Basically, this in-vitro fertilization (IVF) based technique allows the replacement of defective mitochondrial DNA in a woman who carries the disease with healthy mitochondria from another woman. While the baby will still have the nuclear DNA (the vast majority of genes) of the mother and father, it will have the healthy mitochondrial DNA from the donor woman as well. Thus, the baby would not have mitochondrial disease.
Currently, mitochondrial donation is only permitted in the United Kingdom. However, as the process is well-regulated, only people with severe disease have access to the procedure. Other issues include the availability of qualified donors willing to provide immature eggs, as well as evidence that the technique may not be 100% effective. Additionally, some countries may ban mitochondrial donation for ethical reasons.
Families with mitochondrial disease causes by nuclear DNA mutations (as opposed to mitochondrial defects) have preventative options that are associated with other nuclear genetic disorders. These include genetic counseling and prenatal testing (such as amniocentesis).
Prognosis

The outlook for people with mitochondrial disease is highly variable and unpredictable (via a 2014 study published in the journal Acta Myologica). The reason for this is that the course of the disease is determined by a number of factors, including the type of disease and how many organ systems are involved. While early diagnosis and treatment of symptoms improves the prognosis, many children with mitochondrial disease must struggle with severe disabilities and bleak expectations for survival beyond the teenage years. Adults tend to fare better, but may still experience crippling and life-altering changes over a very brief period of time.
The prognosis should be shaped by any severe symptoms or complications resulting from the disease rather than by the type of mitochondrial disease. Outcomes can be optimized by ensuring that all the needs of the patient are considered upon diagnosis. Periodic medical checkups are also important to track how well the patient is coping with the disease.
Currently, potential treatments and cures for mitochondrial diseases are being investigated by research scientists, notes the National Institute of Neurological Disorders and Stroke. For example, in a cell with both normal and defective mitochondria, the healthy mitochondria may be induced to merge with the defective mitochondria and thus take predominance over the cell. Alternatively, interventions may be developed to drive the creation of new mitochondria that proliferate and overwhelm the mutants.